Inorg. Chem. 前沿:自旋-轨道耦合(SOC)如何重塑重金属 PCET 反应的热力学(前沿门户)



在涉及重过渡金属(如铼、铱、铂等)的氧化还原催化过程中,质子耦合电子转移(PCET)是核心步骤。传统研究在计算其热力学参数(如键离解自由能BDFE)时,往往假设自旋-轨道耦合(SOC)的影响可以忽略,或者在低对称性的强配体场中被消除。然而,本文通过实验与理论的严密结合证明,即使在看似简单的有机金属配合物中,SOC对氧化还原电位的影响也可能高达数百毫伏,从而显著改变对催化活性的评估。
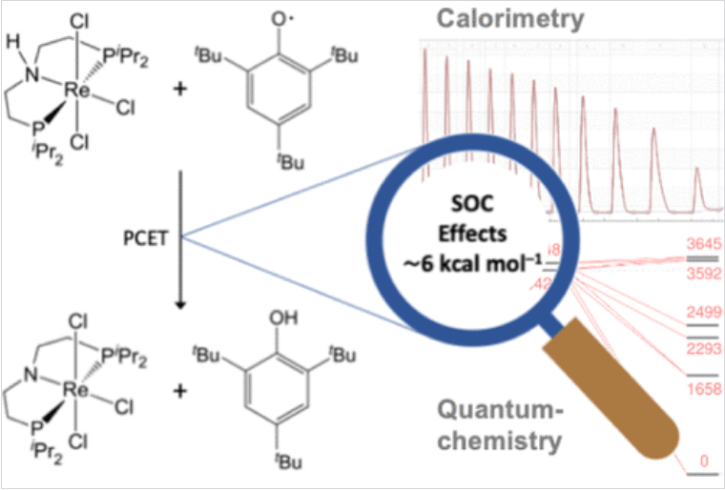
· 核心问题:重过渡金属离子的基态能级经常受到 SOC 的强烈扰动。如果氧化态和还原态受到的 SOC 扰动程度不同,就会在氧化还原电位中引入巨大的偏差。
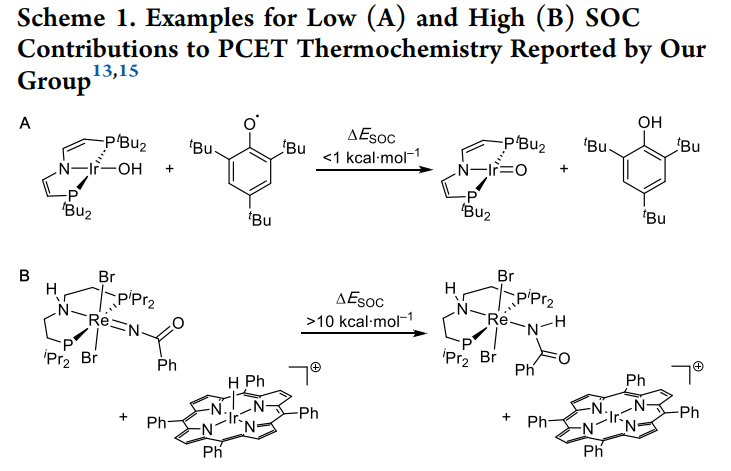
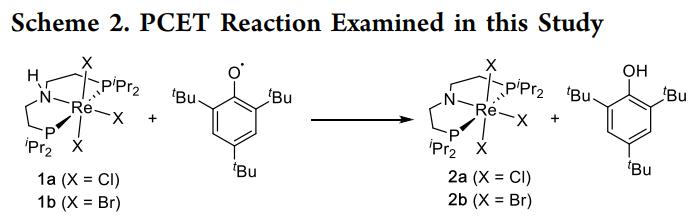
· 研究对象:研究者设计了一个特殊的铼(III)氨基二膦配合物 [ReCl2(PNP)]。该体系可以发生单电子氧化生成铼(IV),或发生 PCET 过程。
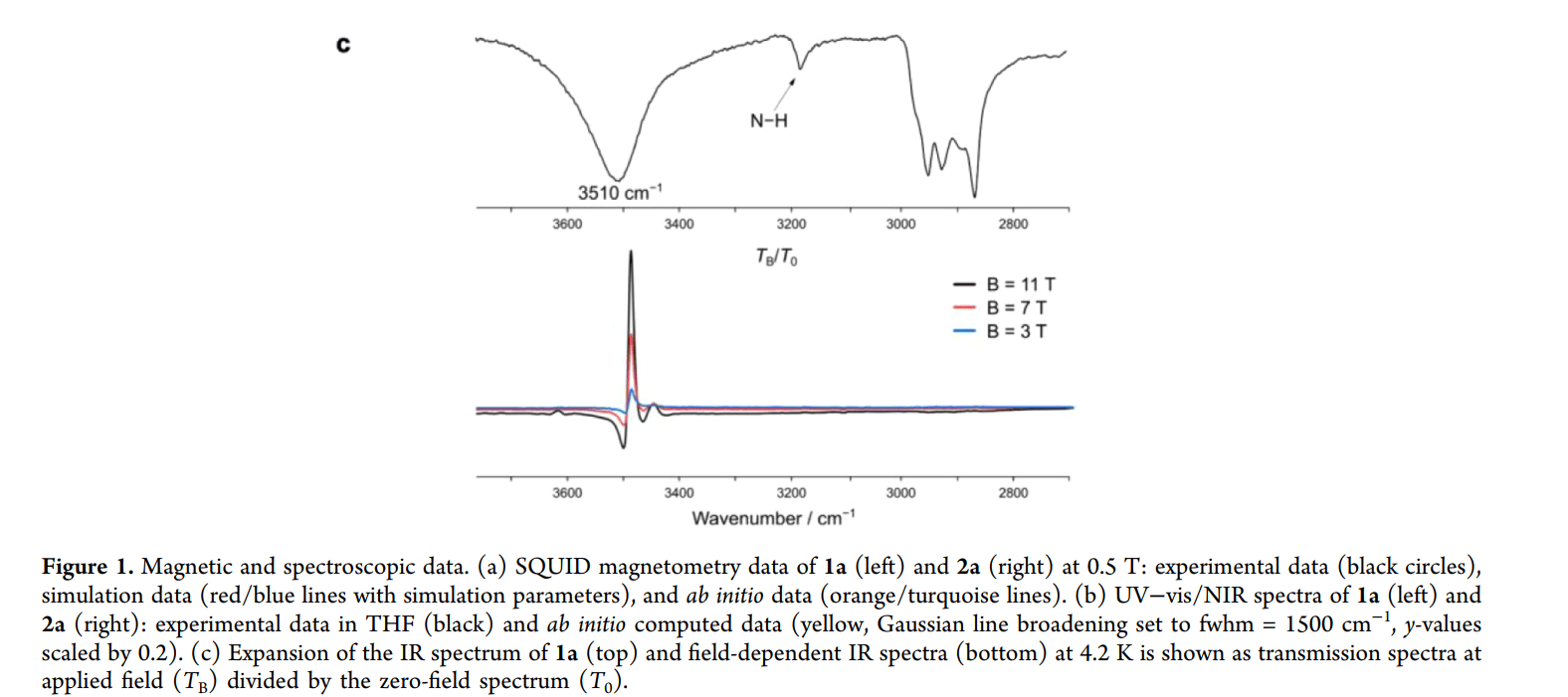
1. 实验热力学测定(精确称量能量)
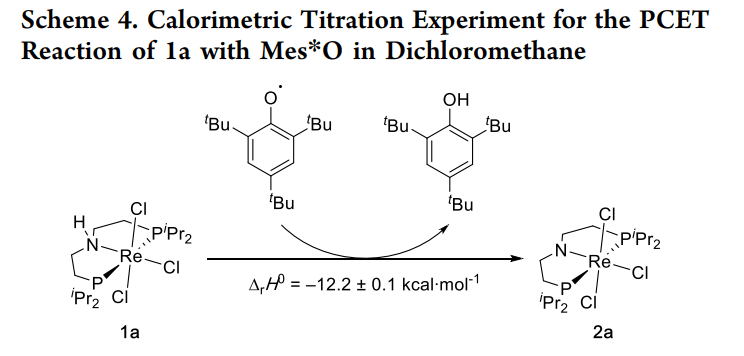
研究者利用等温滴定微热量计(ITC)和平衡常数测定,构建了一个完整的热力学循环:
· 测定了 [Re(III)-NH] 转化为 [Re(IV)-N] 的质子耦合电子转移过程。
· 实验得到的 N-H 键离解自由能(BDFE)与常规 DFT 计算值之间存在明显的“缺口”。
2. DFT 计算细节与能级演变
为了弥补上述“缺口”,研究者进行了精密的计算模拟:
· 计算水平:使用 TPSSh 泛函,结合 def2-TZVP 基组。
· 相对论校正:通过 ORCA 软件在 ZORA(零阶正则近似)水平下处理相对论效应。
· SOC 处理:引入了准简并微扰理论(QDPT)来显式计算自旋-轨道耦合常数。
3. 关键发现:SOC 如何改变结果?
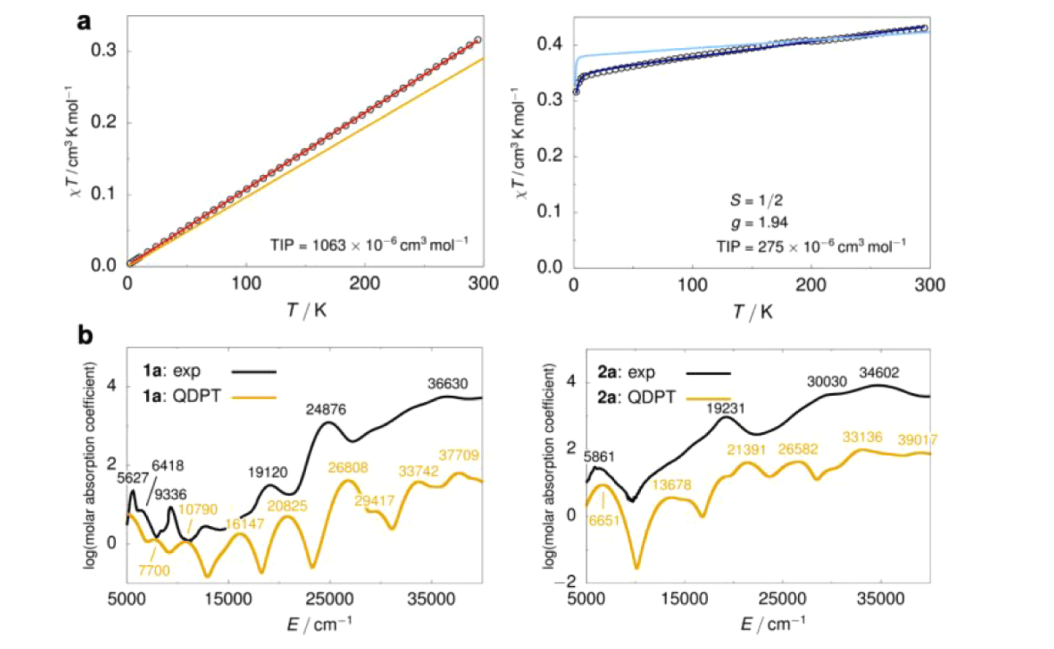
· 基态分裂:对于 d3 构型的 Re(IV),SOC 导致其基态能级发生了显著分裂(零场分裂 ZFS)。
· 能量贡献:计算发现,SOC 对该体系氧化还原电位的贡献约为 0.2 V 至 0.3 V。如果不考虑这一项,计算出的 BDFE 将会产生约 5-7 kcal/mol 的误差。
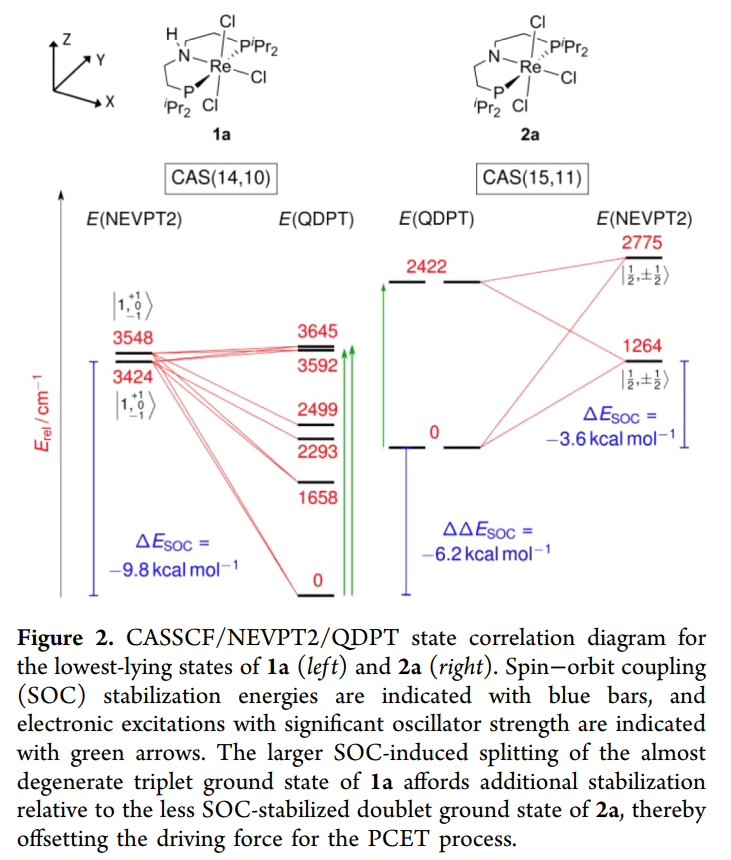
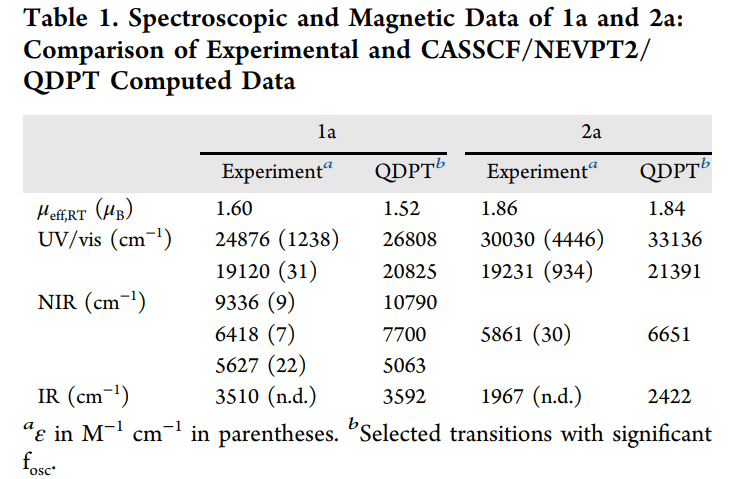
1. 电子排布的非简并性: 在低对称性(如 C2v)的配合物中,d 轨道已经发生分裂。通常认为这会“冻结”轨道角动量,但对于铼这种重元素,其 SOC 常数(zeta)非常大,足以越过轨道能级差产生显著的二级微扰效应。
2. PCET 过程中的状态切换: 从 Re(III) 到 Re(II) 或 Re(IV),金属中心的电子数发生变化,磁性质随之改变。SOC 对不同氧化态的稳定化作用不对等,直接导致了实验观测值的偏移。
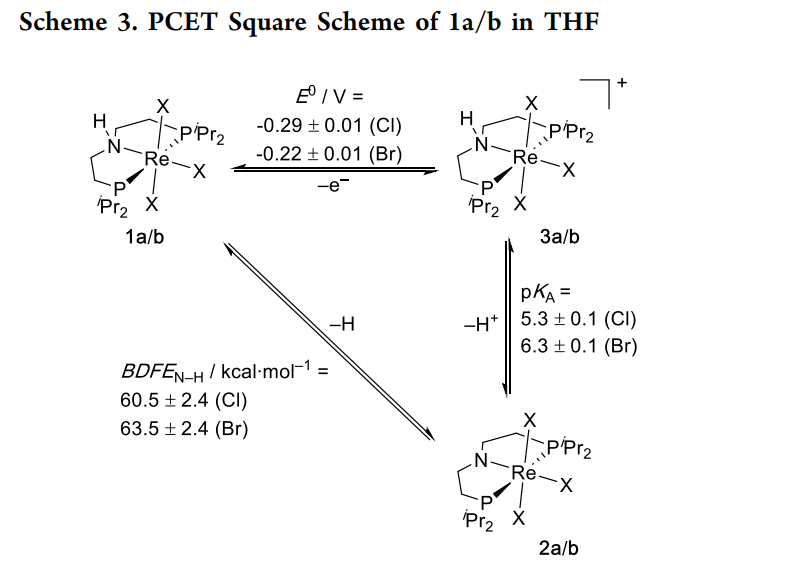
该文献通过铼配合物的 PCET 反应实例,定量评估了自旋-轨道效应(SOC)对热力学参数的影响。研究利用高精度量热实验与考虑相对论效应的 DFT 计算(ZORA-SOC)相互验证,证实了 SOC 能使重金属配合物的氧化还原电位产生数百毫伏的偏移。这一研究强调了在处理重元素催化机理时,必须超越传统的非相对论量子化学框架,将自旋-轨道相互作用纳入核心考虑范畴,为精确模拟重金属催化反应提供了重要范式。








